This web page was produced as an assignment for Genetics 677, an undergraduate course at UW-Madison.
What is Protein Phylogeny?
Phylogeny, in general, is the study of the relatedness of groups of organisms through molecular sequencing and morphological (physical) characteristics. These groups of organisms can be different populations of the same organism, so for example two different populations of humans, or different organisms, so humans and chimpanzees. Protein phylogeny involves comparing protein sequences between groups to infer the relatedness of the protein in question. This information can then be used to construct a phylogenetic tree, as shown in Figure 1 below.
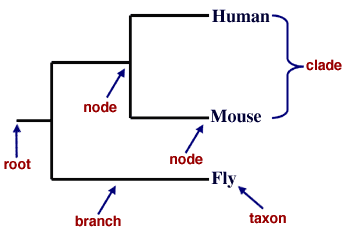
Figure 1. The parts of a phylogenetic tree. The taxa in this tree are "human", "mouse", and "fly." Several nodes are indicated, such as the "fly" taxon node and an internal node that represents the common ancestor of mice and humans. The root is indicated at left, representing the common ancestor of all three taxa listed.
Phylogenetic trees are meant to be a visual representation of evolution. As speciation events occur, branch points form, and as particular organisms become extinct, branches terminate. There has been a great deal of debate on how to classify organisms, and this page will focus solely on the protein sequencing methods.
Below there are four examples of four different types of phylogenetic trees. In order to obtain a neighbor joining tree, it is necessary to know the distance between each sequence you are trying to compare [1]. This tree starts out looking like a star, very disorganized, then the algorithm looks for a pair of sequences that are closest together and creates a new node. The length of the branches represent how far apart the sequences are from one another.
The second is an average distance tree, which requires the proteins in question to be aligned before analysis. For more on FRMD7 alignment, please visit the Protein Homology and Alignments page. Any gap in the alignment is ignored, or counted as a mismatch [2].
The last two trees on this page came from the same program, but one is rooted, and the other is unrooted. The root on the first tree simply represents the most recent common ancestor of the tree, even if this ancestor is extinct or unknown.
Below there are four examples of four different types of phylogenetic trees. In order to obtain a neighbor joining tree, it is necessary to know the distance between each sequence you are trying to compare [1]. This tree starts out looking like a star, very disorganized, then the algorithm looks for a pair of sequences that are closest together and creates a new node. The length of the branches represent how far apart the sequences are from one another.
The second is an average distance tree, which requires the proteins in question to be aligned before analysis. For more on FRMD7 alignment, please visit the Protein Homology and Alignments page. Any gap in the alignment is ignored, or counted as a mismatch [2].
The last two trees on this page came from the same program, but one is rooted, and the other is unrooted. The root on the first tree simply represents the most recent common ancestor of the tree, even if this ancestor is extinct or unknown.
Human FRMD7 Protein Phylogeny
Human FRMD7 protein was compared to six different species - dog, cattle, rat, chicken, cattle, Drosophila melanogaster (fruit flies), and C. elegans - in order to obtain the phylogenetic trees below. Data was obtained through ClustalW2 and Phylogeny.fr which uses the MUSCLE program to perform analysis.
ClustalW2 Alignment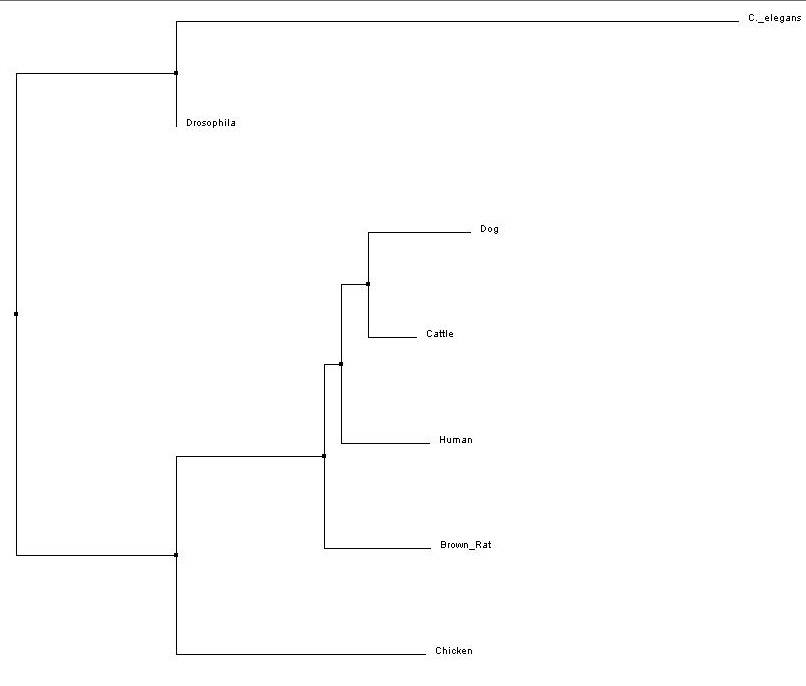
Figure 2. Neighbor Joining (Click to Enlarge)
|
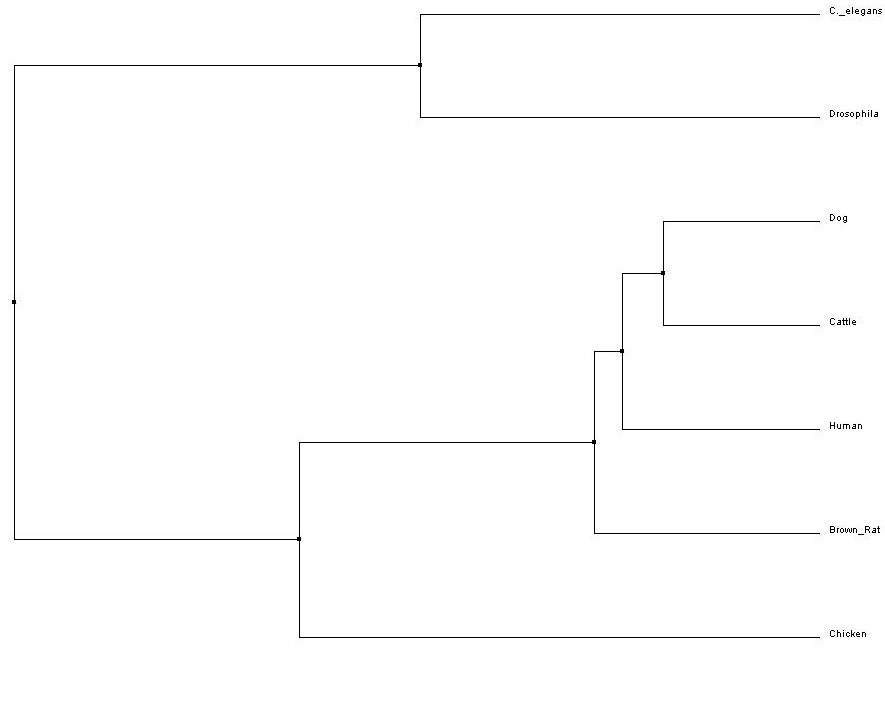
Figure 3. Average Distance (Click to Enlarge)
|
Phylogeny.fr Alignment Via MUSCLE [3, 4, 5, 6, 7, 8, 9]
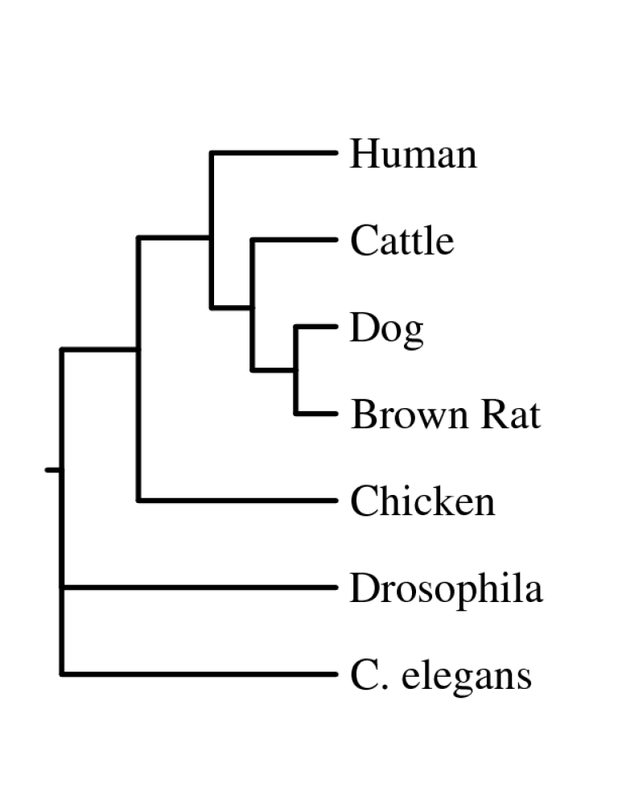
Figure 4. Drawgram Rooted (Click to Enlarge)
|

Figure 5. TreeDyn Unrooted (Click to Enlarge)
|
Analysis and Discussion
All four trees show Drosophila and C. elegans as an outgroup, which suggests those two organisms have proteins that are less closely related to the other five that were analyzed - human, cattle, dog, rat and chicken. The neighbor-joining and average distance trees obtained from ClustalW2 are nearly identical, and show human FRMD7 protein to be closely related to cattle and dog FRMD7 protein. This differs from the tress obtained from Phylogeny.fr, which show human FRMD7 protein to be more closely related to the cattle FRMD7, then the dog FRMD7. This is most likely due to the different programs used by ClustalW2 and Phylogeny.fr.
References
[1] Didelot, X., Robinson D. A., Falush, D., Feil E. J., (2010).
Sequence-based analysis of bacterial population structures. Bacterial Population Genetics in Infectious Disease 2010(Ch. 3) 46. ISBN: 9780470424742. Retrieved from: http://books.google.com/books?id=gPVjfsWnGCcC&pg=PA46#v=onepage&q&f=false
[2] Mount , D. M. (2004)
Bioinformatics: Sequence and Genome Analysis 2nd ed. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY
[3] Dereeper A., Audic S., Claverie J. M., Blanc G. (2010)
BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BioMed Central Evolutionary Biology, 2010(10:8). doi: 10.1186/1471-2148-10-8
[4] Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.F., Guindon S., Lefort V., Lescot M., Claverie J.M., Gascuel O. (2008)
Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Research. 2008(1;36) W465. doi: 10.1093/nar/gkn180
[5] Edgar R. C. (2004)
MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004(32:5), 1792. doi: 10.1093/nar/gkh340
[6] Castresana J. (2000)
Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution, 2000(17), 540. Retrieved from: http://mbe.oxfordjournals.org/content/17/4/540.long
[7] Guindon S., Gascuel O. (2003)
A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systems Biology. 2003(52), 696. doi: 10.1080/10635150390235520
[8] Anisimova M., Gascuel O. (2006)
Approximate likelihood ratio test for branchs: A fast, accurate and powerful alternative. Systems Biology. 2006(55), 539. doi: 10.1080/10635150600755453
[9] Felsenstein J. (1989)
PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics, 1989(5), 164. Retrieved from: http://www.citeulike.org/user/rvosa/article/2346707
Sequence-based analysis of bacterial population structures. Bacterial Population Genetics in Infectious Disease 2010(Ch. 3) 46. ISBN: 9780470424742. Retrieved from: http://books.google.com/books?id=gPVjfsWnGCcC&pg=PA46#v=onepage&q&f=false
[2] Mount , D. M. (2004)
Bioinformatics: Sequence and Genome Analysis 2nd ed. Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY
[3] Dereeper A., Audic S., Claverie J. M., Blanc G. (2010)
BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BioMed Central Evolutionary Biology, 2010(10:8). doi: 10.1186/1471-2148-10-8
[4] Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.F., Guindon S., Lefort V., Lescot M., Claverie J.M., Gascuel O. (2008)
Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Research. 2008(1;36) W465. doi: 10.1093/nar/gkn180
[5] Edgar R. C. (2004)
MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004(32:5), 1792. doi: 10.1093/nar/gkh340
[6] Castresana J. (2000)
Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Molecular Biology and Evolution, 2000(17), 540. Retrieved from: http://mbe.oxfordjournals.org/content/17/4/540.long
[7] Guindon S., Gascuel O. (2003)
A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systems Biology. 2003(52), 696. doi: 10.1080/10635150390235520
[8] Anisimova M., Gascuel O. (2006)
Approximate likelihood ratio test for branchs: A fast, accurate and powerful alternative. Systems Biology. 2006(55), 539. doi: 10.1080/10635150600755453
[9] Felsenstein J. (1989)
PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics, 1989(5), 164. Retrieved from: http://www.citeulike.org/user/rvosa/article/2346707
Site created by: Kristen Klimo
Last updated: 5/11/2012
University of Wisconsin-Madison
Last updated: 5/11/2012
University of Wisconsin-Madison
